

PATIENTS
Benefiting from the most efficient or adapted therapy to cure from disease and/or to be cured with the best quality of life during treatment
ACOBIOM
Providing innovative and non-invasive Blood-based In Vitro Diagnostic to predict patient response to a certain therapy
CLINICS & HOSPITALS
Making a diagnosis, treating, providing medical follow-up with the objective to personalize patient’s medical care in taking into account individual differences
GemciTest ®
The first diagnostic to predict the patient response in pancreatic cancer treatment
PANCREATIC CANCER
A rare and “silent” disease with no symptom early on, difficult to diagnose and cure.
DIAGNOSTIC
A blood-based molecular test (RUO diagnostic) to predict patient’s therapeutic response
GEMCITABINE
The less toxic and expensive chemotherapy in pancreatic cancer treatment
BLOOD RNA-SIGNATURE
A combination of biomarkers to identify patients most likely to benefit from gemcitabine
Prescribing the right treatment to the right patient requires funds.
To financially support ACOBIOM’s developments, please visit the “Fundraising” page
Our Expertises
Acobiom is an innovative biotechnology company specialized in the discovery of new biomarkers and in the development of innovative diagnostics.
BIOMARKERS
Analyses of biological phenotypes; investigation of gene expression changes; Identification & validation of acid nucleic (DNA, RNA, miRNA, small RNA, coding & non coding RNA) biomarkers based on Sequencing/NGS & PCR technologies.
BIOINFORMATICS
Treatments & analyses of sequencing/omics big data through a proprietary suite of Bioinformatic and Biostatistic tools; Development of a 21,000 Human RNA-Seq database, in-silico investigations through public & proprietary databases.
PRECISION MEDICINE
Biomarkers and diagnostics “to optimize a specific preventive, diagnostic or therapeutic intervention in a given subpopulation of patients, which would most likely to benefit from it”.
GENOMICS
A unique scientific and technical expertise associated with 20-year experience in genomics & transcriptomics, in sequencing/NGS & PCR technologies, in performing RNA-Seq / ChIP-seq analyses, and PCR / qPCR / qRT-PCR experiences.
Latest News
The Intricate Interplay between Innate Immunity and Pancreatic Cancer
Innate immunity’s role in pancreatic cancer is complex. It impacts inflammation, immune surveillance, and the tumor microenvironment, influencing disease progression and potential treatments.
Read more
A new diagnostic to predict the patient response to FOLFIRINOX in pancreatic cancer
Innovation in pancreatic cancer: Acobiom develops the 1st blood-based diagnostic to identify the patients most likely to benefit from FOLFIRINOX as 1st-line treatment.
Read more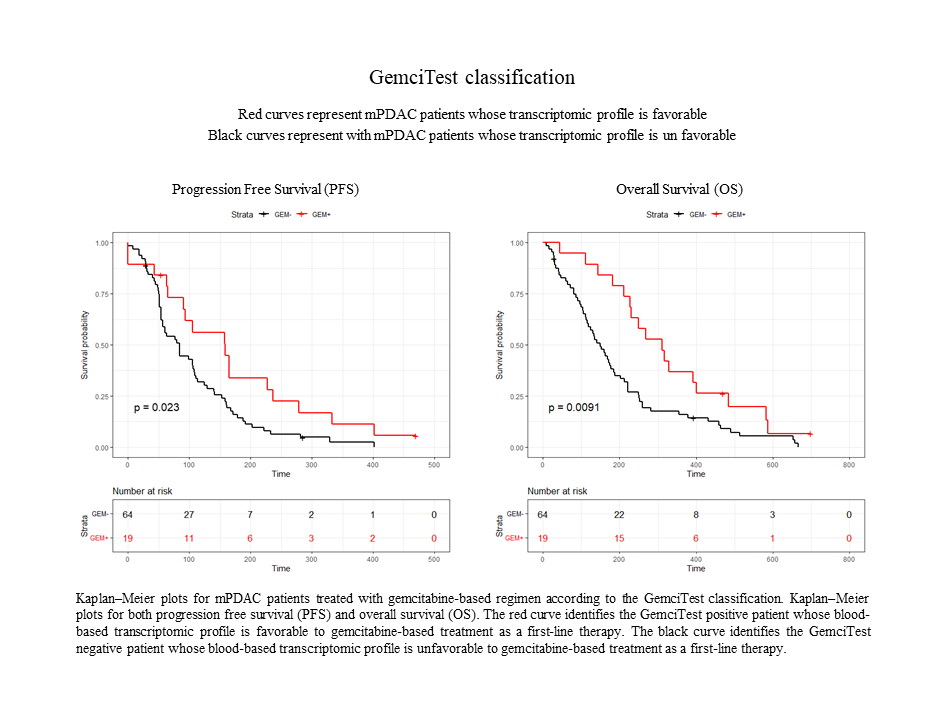
Performance of a blood-based RNA signature for gemcitabine-based treatment in metastatic pancreatic adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC, representing more than 90% of pancreatic cancer cases is a highly lethal cancer, and chemotherapy is a key treatment for advanced PDAC. Gemcitabine chemotherapy is still[…]
Read moreABOUT ACOBIOM
Since its foundation, Acobiom has always been at the forefront of Medical Innovation.
Acobiom was founded by Didier Ritter and Dr David Piquemal under the name « Skuldtech » to promote the gene expression profile (transcriptome) approach with the objective to discover RNA biomarkers, to develop diagnostics, and to improve the patient’s medical care.
FOR MORE INFORMATION

